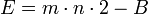Интернет магазин китайских планшетных компьютеров |
|
Компьютеры - BLAST - Принципы работы BLAST29 мая 2011Оглавление: 1. BLAST 2. Принципы работы BLAST Все выравнивания принято делить на глобальные и локальные. Программы серии BLAST производят локальные выравнивания, что связано с наличием в различных белках сходных доменов и паттернов. Кроме этого локальное выравнивание позволяет сравнить иРНК с геномной ДНК. В случае глобального выравнивания обнаруживается меньшее сходство последовательностей, особенно их доменов и паттернов. После введения изучаемой нуклеотидной или аминокислотной последовательности на одну из веб-страниц BLAST, она вместе с другой входной информацией, значение величины E и др.) поступает на сервер. BLAST создаёт таблицу всех «слов» и сходных «слов». Затем в базе данных проводится их поиск. Когда обнаруживается соответствие, то делается попытка продлить размеры «слова» сначала без гэпов, а затем с их использованием. После максимального продления размеров всех возможных «слов» изучаемой последовательности, определяются выравнивания с максимальным количеством совпадений для каждой пары запрос — последовательность базы данных, и полученная информация фиксируется в структуре SeqAlign. Форматер, расположенный на сервере BLAST, использует информацию из SeqAlign и представляет её различными способами. Для каждой обнаруженной в базе данных программами BLAST последовательности необходимо определить, насколько она сходна с изучаемой последовательностью и значимо ли это сходство. Для этого BLAST вычисляет число битов и величину Е для каждой пары последовательностей. При определении сходства ключевым элементом является матрица замен, так как она определяет показатели сходства для любой возможной пары нуклеотидов или аминокислот. В большинстве программ серии BLAST используется матрица BLOSUM62. Исключением являются blastn и megablast. С помощью модифицированных алгоритмов Смита-Уотермана или Селлерса определяются все пары сегментов, которые нельзя увеличить, так как это приведёт к уменьшению показателей сходства. Такие пары продленных «слов» называются парами сегментов с максимальным сходством. В случае достаточно большой длины изучаемой последовательностей и последовательности базы данных показатели сходства HSP характеризуются двумя параметрами K и P. Эти показатели необходимо указывать при приведении показателей сходства изучаемой последовательности и последовательности базы данных. Для сравнения показателей сходства различных выравниваний независимо от используемой матрицы, их необходимо преобразовать. Для получения преобразованного показателя сходства используют формулу: Величина B показывает, насколько сходны последовательности. Так как в формулу расчёта B заложены показатели К и P, то нет необходимости указывать их при приведении значений B. Величина E, соответствующая показателю B, показывает достоверность данного выравнивания. Она определяется по формуле: Программы BLAST преимущественно определяют значение E, а не P. Но при E < 0,01 значения P и E почти идентичны. Величина E определяется по формуле при сравнении лишь двух аминокислотных или нуклеотидных последовательностей. Сравнение изучаемой последовательности длиной m с множеством последовательностей базы данных может основываться на двух положениях. Первое положение состоит в том, что все последовательности базы данных одинаково сходны с изучаемой. Это подразумевает, что значение E для выравнивания с короткой последовательностью, содержащейся в базе данных, следует приравнять со значением E для выравнивания с длинной последовательностью. Для вычисления значения E по базе данных необходимо умножить значение E, полученное при попарном сравнении, на число последовательностей в ней. Второе положение заключается в том, что изучаемая последовательность более сходна с короткими, а не с длинными последовательностями, потому что последние часто состоят из различных участков. Если предположить, что вероятность сходства пропорциональна длине последовательности, то попарное значение E для последовательности базы данных длиной n надо умножить на N/n, где N — общая длина аминокислот или нуклеотидов в базе данных. Программы BLAST преимущественно используют этот подход для вычисления значений E по базе данных. Теоретически локальное выравнивание может начинаться с любой пары нуклеотидов или аминокислот выровненных последовательностей. Однако HPS, как правило, не начинаются близко к краю последовательностей. Для коррекции такого краевого эффекта необходимо вычислять эффективную длину последовательностей. В случае последовательностей длиной более 200 остатков происходит нейтрализация краевого эффекта. Просмотров: 2190
|